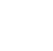Sample Processing and Analysis
Sample processing options encompass the areas of moisture management, particle size selection, particle size reduction, and sample digestion or extraction. The project team should choose the most appropriate processing options for a specific site characterization. This section includes implementation guidance for the various processing options recommendations for QC.
5.1 Introduction
Section 2 discusses some of the common sources of sampling uncertainty, but to obtain representative samples, sampling uncertainty must be limited or managed (Ramsey and Hewitt 2005). In the absence of uncertainty, a sample result by definition would be accurate. However, it is impossible to completely eliminate uncertainty and produce an accurate result unless all the soil in the DU is included in the analytical determination, which is obviously impractical. Thus, limiting sampling uncertainty is a critical function of any sampling design, implementation, processing, and analysis.
Incremental sampling has been successfully implemented at numerous sites for a variety of contaminants, and multiple processing options are available depending on the contaminants (such as energetics, metals, perchlorate, white phosphorus, SVOCs, VOCs, PCBs, and so on). Prior to implementation of sample processing, the entire sample collected in the field should be shipped to the laboratory. There may be some situations when it is desired to perform some of the sample processing steps in the field, such as air-drying and sieving, but the preferred approach is to conduct this activity in a controlled environmental such as an analytical laboratory.
ISM sample processing techniques, such as milling and representative subsampling, are designed to ensure the (typically small) mass of sample analyzed by the laboratory is representative of the field sample and hence the DU from which it was collected. These techniques reduce data variability as compared with conventional sample handling and processing approaches, but they also introduce some amount of sampling uncertainty. It is recommended that all ISM sample processing be performed in a controlled laboratory setting to minimize this sampling uncertainty. However, depending on site logistics, type of soil, total number and/or mass of ISM samples, and so on, sample processing can be initiated in the field for some analytes (such as SVOCs, pesticides, PCBs, and metals) with appropriate cautions as noted below. The techniques available include air-drying, disaggregation, sieving, milling, subsampling, and digestion or extraction.
5.2 Choosing Appropriate ISM Processing Options
The project team should choose the most appropriate sample processing options for a specific site characterization since there is no single combination of processing techniques applicable to all sites. Site characterization objectives should drive the selection of sample processing options, but there are four general areas to consider: moisture management, particle size selection, particle size reduction, and sample digestion/extraction. Objectives and site characteristics such as COPCs (analytes), surface or subsurface soil, contaminant release mechanisms, and data usage scenarios can also influence the selection of sample processing options.
There are also three important questions that must be answered to select the most appropriate sample processing options: Is air-drying acceptable? Is particle size selection needed? Is particle size reduction appropriate?
The specific analytes must be decided upon before finalizing sample processing decisions. There can be a wide range of physical and chemical characteristics within analyte groups, and these differences can influence the selection of sample processing options. Consider characteristics such as boiling point, volatility, air reactivity, sorption of analyte to particles, and the presence of high-concentration nuggets. Other considerations include particle size, distribution of analytes on or in the particles, particle weathering, contaminant release mechanism(s), and the end use of the data.
5.2.1 Air-drying
Moist samples may need to be air-dried until the soil aggregates are crushable to facilitate disaggregation and sieving, but drying to constant weight is not necessary for this purpose. The samples should be dried in a dust-free location where temperatures and UV light are not expected to cause degradation of COPCs. Air-drying is appropriate if the analytes are chemically stable when exposed to air and have sufficiently high boiling points or are strongly sorbed to the particles such that they are unlikely to volatilize during extended air exposure at the selected drying temperature. Drying at ambient temperature (15 to 25°C) is most common and may take up to several days, thus impacting turnaround time and remaining holding time. Elevated temperature drying (30 to 105°C) accelerates the drying process but also requires greater analyte stability. The binding (distribution coefficient, soil organic carbon-water partitioning coefficient) between the contaminant and the soil particle should also be considered. Air-drying can be acceptable for strongly absorbed, low boiling point analytes, which would be expected for weathered surface soils.
Table 5-1 lists several example explosives and SVOCs, their boiling points, and their estimated loss potential during the room temperature air-drying step when these analytes are weakly sorbed to the soil matrix (Bruce 2003). Air-drying produces crushable soil particles, but it risks loss of low boiling point target analytes. Table 5-1 is not all-inclusive and is intended only as an example for evaluating COPCs and the possible effects of air-drying. Physical property data for additional COPCs are available in “Technical Guidance Manual Notes: Decision Unit and Multi-Increment Sample Investigations,”Tables 2a and 2b (HDOH 2011).{HDOH, 2003 #254;HDOH, 2011 #254} Applying air-drying to analytes that are not significantly weathered with moderate and large loss risks should be avoided unless there is sufficient site knowledge or experimental data to demonstrate the loss risk is acceptable.
Table 5-1. Potential for loss during the air-drying step for weakly sorbed analytes.
Source: ITRC ISM-1 Team, 2012.
| Contaminant | Vapor Pressure (mm Hg) |
Boiling Point (℃) |
Loss Potential |
| Acenaphthene | 2.15E-03 | 279 | Moderate |
| Acenaphthylene | 6.68E-03 | 280 | Moderate |
| 2-Amino-4,6-dinitrotoluene | 3.33E-06 | 352 | Small |
| 4-Amino-2,6-dinitrotoluene | 3.65E-06 | 352 | Small |
| bis(2-Chloroethoxy)methane | 1.32E-01 | 218 | Small |
| bis(2-Chloroethyl)ether | 1.55E+00 | 179 | Moderate |
| bis(2-Chloro-1-methylethyl)ether | 5.60E-01 | 187 | Moderate |
| 4-Chloro-3-methylphenol | 5.00E-02 | 235 | Moderate |
| 2-Chloronaphthalene | 1.22E-02 | 256 | Moderate |
| 2-Chlorophenol | 2.53E+00 | 175 | Moderate |
| Dibenzofuran | 2.48E-03 | 287 | Small |
| 1,2-Dichlorobenzene | 1.47E+00 | 180 | Large |
| 1,3-Dichlorobenzene | 2.15E+00 | 173 | Large |
| 1,4-Dichlorobenzene | 1.74E+00 | 174 | Large |
| 2,4-Dichlorophenol | 9.00E-02 | 210 | Small |
| Dimethylphthalate | 3.08E-03 | 284 | Small |
| 1,2-Dinitrobenzene | 4.55E-05 | 318 | Small |
| 1,3-Dinitrobenzene | 9.00E-04 | 291 | Small |
| 2,4-Dinitrotoluene | 1.47E-04 | 300 | Small |
| 2,6-Dinitrotoluene | 5.67E-04 | 300 | Small |
| Hexachlorobutadiene | 2.20E-01 | 215 | Large |
| Hexachloroethane | 2.10E-01 | 154 | Large |
| Octahydro-1,3,5,7-tetranitro-1,3,5,7- tetrazocine (HMX) | 3.30E-14 | 436 | Small |
| Isophorone | 4.38E-01 | 215 | Large |
| 2-Methylnaphthalene | 5.50E-02 | 241 | Moderate |
| 4-Methylphenol | 1.10E-01 | 202 | Moderate |
| Naphthalene | 8.50E-02 | 218 | Large |
| Nitrobenzene | 2.45E-01 | 211 | Large |
| Nitroglycerin | 4.00E-04 | 250 | Small |
| N-Nitrosodimethylamine | 2.7E+00 | 154 | Moderate |
| N-Nitroso-di-n-propylamine | 3.89E-01 | 206 | Small |
| 2-Nitrotoluene | 1.88E-01 | 222 | Moderate |
| 3-Nitrotoluene | 2.05E-01 | 232 | Moderate |
| 4-Nitrotoluene | 1.57E-02 | 238 | Moderate |
| Pentaerythritol tetranitrate (PETN) | 5.45E-09 | 364 | Small |
| Phenol | 3.50E-01 | 182 | Small |
| Hexahydro-1,3,5-trinitro-1,3,5- triazine (RDX) | 4.10E-09 | 353 | Small |
| Methyl-2,4,6-trinitrophenylnitramine (Tetryl) | 1.17E-07 | 432 | Moderate |
| 1,2,4-Trichlorobenzene | 4.60E-01 | 214 | Large |
| 2,4,6-Trichlorophenol | 8.00E-03 | 246 | Small |
| 1,3,5-Trinitrobenzene | 6.44E-06 | 315 | Moderate |
| 2,4,6-Trinitrotoluene | 8.02E-06 | 365 | Small |
Considering analyte loss risks during the air-drying step, it may be necessary on occasion to skip air-drying and proceed with other processing steps on the as-received sample, typically when lower boiling point SVOCs are primary contaminants. HDOH has also expressed concerns about the potential loss of elemental mercury. Wet, sticky samples cause mechanical problems when processing, but 2D slabcake subsampling on the as-received sample is possible when air-drying must be avoided. Note that decreased reproducibility occurs as other processing steps are skipped.
In most cases, air-drying is performed to facilitate disaggregation, sieving, milling, and extraction. When low boiling analytes are weakly retained on the soil particles, they can be lost during the air-drying step and produce a low bias in the final results. This is generally not a concern for surface soil samples unless the COC release is very recent. At most sites, the surface soil has been exposed to air for months, years, or decades since the release. Thus, air exposure at the laboratory for a few days during air-drying is not likely to produce analyte loss for surface soils. Figure 5-1 shows that low boiling PAHs are not lost during air-drying from an aged surface soil. Three replicate subsamples were collected immediately when the soil was spread out for air-drying and after processing (drying, disaggregation, and sieving). The average results demonstrate no analyte loss from surface soil samples during air-drying and that precision is generally improved.
Conversely, recent releases and subsurface soil samples might produce circumstances where low boiling analytes are weakly sorbed on the particles and are at risk for loss during air-drying. Subsampling of the as-received sample using the 2D slabcake process is the most common accommodation, but wet sieving and wet milling are viable options if the sample is sufficiently flowable (high water content sediments). As a general recommendation, drying should be utilized only to the extent necessary to avoid potential analyte loss. Moisture content below 5 to 10% is usually acceptable to produce crushable soil aggregates.
5.2.2 Disaggregation prior to other processing
Sample disaggregation is a technique used on dry, crushable soil to break up the aggregates formed during air-drying, but it does not mill small pebbles and other hard particles into smaller particulates, like the particle size reduction techniques (such as milling) listed below. In some risk assessment scenarios, disaggregation is preferable to milling because some metallic COPCs remain “locked” inside the hard particles and are not included in subsequent analyses.
5.2.3 Particle size selection: sieving
Particle size selection and particle size reduction decisions are determined by the DQOs. If the characterization uses an operational soil definition of <2-mm particles, then disaggregation followed by particle size selection with a #10 sieve is a common choice. For the skin-to-mouth and hand-to-mouth exposure pathway, USEPA recommends that soil Pb be measured on the <0.15-mm soil fraction to estimate EPC since this is the particle size most likely to stick to a child’s hands and enter homes as dust (USEPA 2016). Only the particle fraction passing through a 100-mesh sieve is to be analyzed. Other data usage scenarios will push toward other sieve sizes or determine that no sieving is appropriate. Example guidance on sieve selection is available in multiple references (DOD/DOE 2018) (USEPA 2013) (Frederick, Frame, and Vallero 2017, USEPA 2011) (USEPA 2012b) (HDOH 2016b, 2015).
Occasionally, the sample container includes objects not considered part of the sample, so the DQOs should direct whether vegetation and oversized material are included or excluded from the sample. If the Toxicity Characteristic Leaching Procedure(TCLP)is going to be performed on the sample, all material must be retained. Vegetation and oversized material can be manually removed with tweezers or spatulas but can be removed more reproducibly with a sieve if the sample is dried. The excluded materials can be documented via photographs and weight removed when appropriate. Note that in the case of energetic materials and other situations where the COPC is deposited on the ground surface as a particulate, the vegetation should not be removed in the field prior to laboratory sieving.
Although sieving to the <2-mm particle size is typical, there may be contaminant investigations or analyses where alternative particle sizes may be of interest. In these cases, the rationale for sieving to other specific particle sizes and associated changes to laboratory processing/analysis should be clearly discussed during the project planning step and documented in the QAPPs.
5.2.4 Particle size reduction: milling
If a COPC is present as a solid particulate, particle size reduction through sample milling can facilitate more representative subsampling by reducing the range of particle sizes and the maximum size present. The selection of the particle size reduction technique and maximum target particle size should be determined during project planning and is part of DQO development. It should be noted that the maximum particle size has a significant effect on FE. See the discussion in Section 2.6.2.1 on Gy’s TOS about the relationships among particle size, uncertainty, subsample size, and FE. Selection of the appropriate milling process and equipment to achieve the maximum particle size is determined during project planning (see Section 3.1), with many common options described in USEPA guidance (Gerlach and Nocerino 2003). Examples of when particle size reduction may be appropriate after particle size selection include, but are not limited to, metals at small-arms ranges, clay target fragments at skeet ranges, lead-based paint chips, munitions constituents, and so on.
Milling is recommended for ISM metals analyses, especially for those situations where the particulate form is present (Clausen et al. 2018a, Clausen, Georgian, and Bednar 2013, Clausen et al. 2013) (Clausen, Georgian, and Bednar 2013, Clausen et al. 2016, Clausen et al. 2018c, b). Subsampling that uses a larger subsample mass in lieu of milling should reduce FE, but in some situations, a larger subsample mass increases the measured variability (relative to small subsamples). Smaller subsamples (1 g) can be biased against including larger particles, thus producing artificially low relative SDs (RSDs).
Milling is not universally recommended for organiccontaminants other than energetics (SW-846 Method 8330B) (USEPA 2006c) although it has been demonstrated to improve precision for PAHs on skeet ranges and PCBs. Volatilization loss of organic COPCs may occur due to increased temperatures during milling, and excessive milling can lead to destruction of organic contaminants, as demonstrated in the mechanochemical dehalogenation (or mechanochemical destruction) soil remediation process. See “Reference Guide to Non-Combustion Technologies for Remediation of Persistent Organic Pollutants in Stockpiles and Soil” (USEPA 2005a) for additional information.
The usefulness of particle size reduction by milling for organic COPCs is usually small because the larger mass (10 to 30 g or more) normally extracted and analyzed and the particulate nugget effect are often minimal. However, nuggets can and do occur for specific organic COPCs – for example, soil analyzed for PAHs at skeet ranges can exhibit a nugget effect due to the deposition of clay pigeon fragments. In such cases, the advantages and limitations of milling for organic COPCs should be evaluated during project-specific systematic planning.
Particle size reduction has been called milling and grinding, but grinding is also used to refer to disaggregation. When using the term grinding, specify the equipment to be used to help ensure accurate communication.
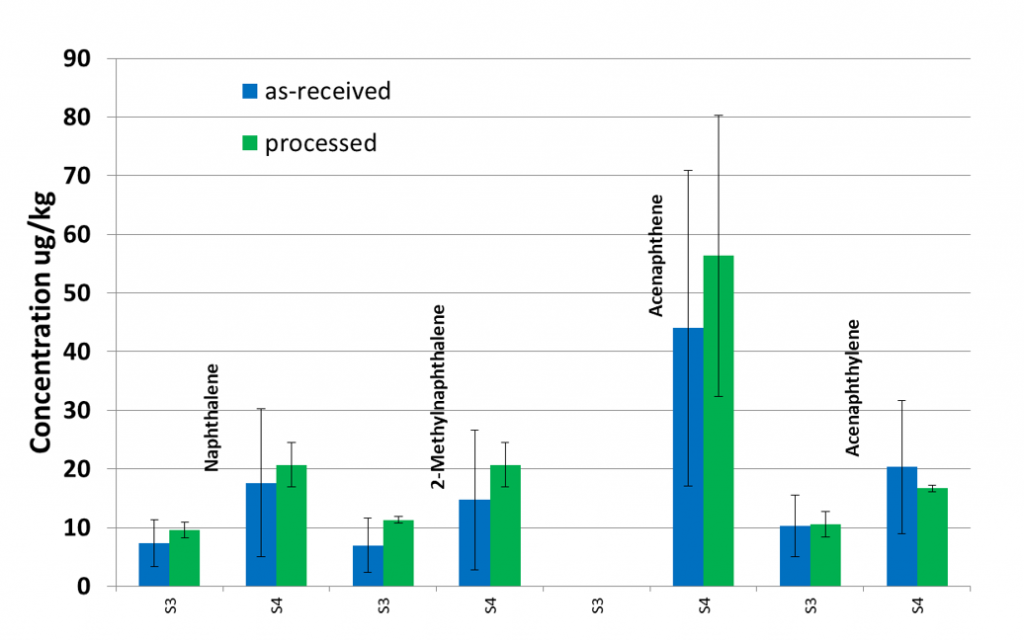
Particle size reduction is generally performed to improve the reproducibility of subsampling (Figure 5-2). More aggressive or longer milling typically produces more improvement to precision.
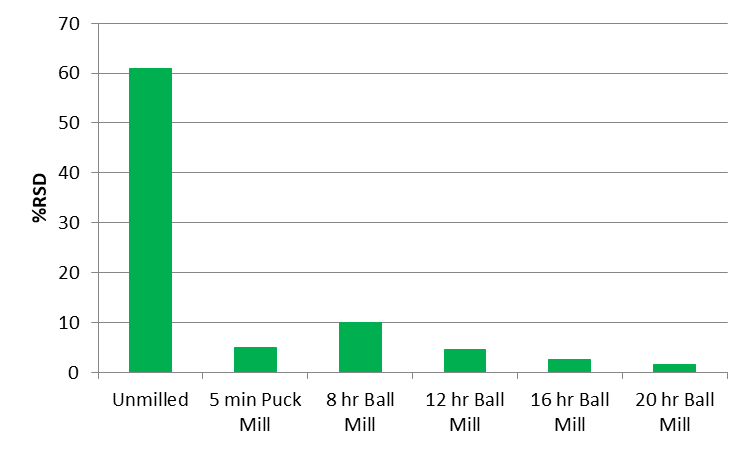
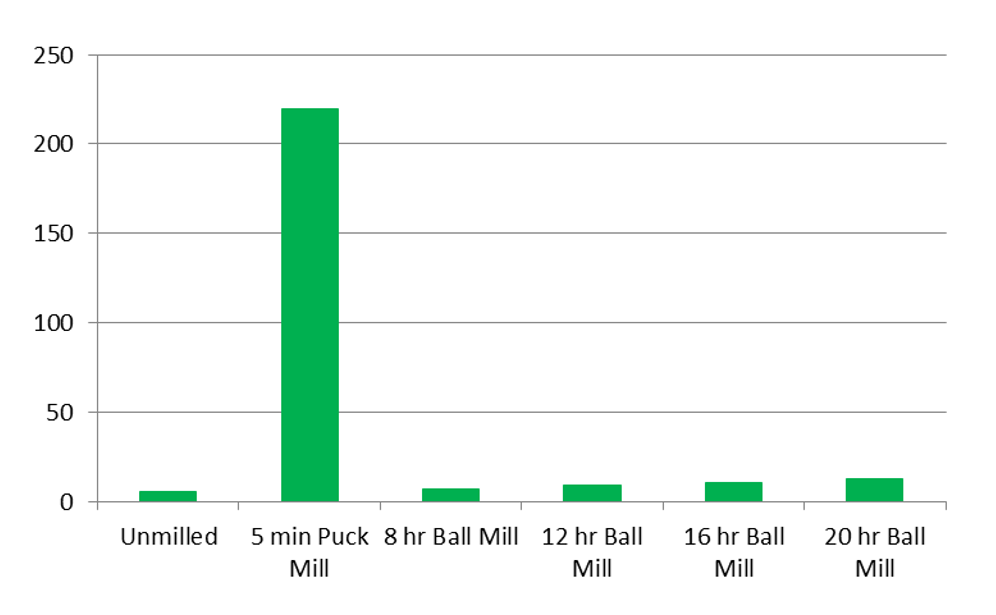
Milling can also be used to intentionally expose the interior of particles to the subsequent digestion or extraction steps. In some instances, this will increase recovery of COCs previously sequestered in the interior of the particles. Figure 5-3 shows this effect for lead particles reduced in size by puck mill and ball mill. In some instances, the increases of the non-anthropogenic metals are negligible and offset by the improved precision and accuracy of the measured metal content (Clausen et al. 2016). The potential improvement in precision and increase in measured metals concentrations should be considered during project-specific systematic planning when determining if milling is appropriate.
Source: Mark Bruce, Eurofins, 2019. Used with permission.
Source: Jay L. Clausen, et al., Microchemical Journal 154 (2020). Used with permission.
Milling can also transfer material from the mill to the sample, which means that mill surfaces with COPCs may interfere with the analysis and should be avoided. If metals are COPCs, then the composition of any metal-containing mill surfaces should be compared with the target analyte list. Ceramic, agate, tungsten carbide, or low chromium steel milling components would be more appropriate. Figure 5-4 shows chromium concentration increasing after the use of a steel puck mill and less contamination from a ball mill with steel can and ceramic grinding media. The use of a milling blank can be useful when a soil-like material with acceptably low background is available. See Section 5.4 for more details.
Source: Mark Bruce, Eurofins, 2019. Used with permission.
Puck mills and ball mills might not be effective at efficiently reducing particle size when the starting size is >2 mm. The use of a jaw crusher can produce particles <2 mm, and for some DQOs, this is sufficient. In other instances, these smaller particles are further particle size reduced in a mill.
5.2.5 Subsampling (2D slabcake on dry or wet samples, sectorial splitter)
Subsampling to produce the analytical samples for extraction, digestion, or leaching is most frequently produced by using the 2D slabcake process. This miniaturized version of what is done in the field is usually a good balance of representativeness and cost. The 1D slabcake can also be used by itself to collect large subsamples or in combination with 2D slabcake to produce small subsamples. The rotary sectorial splitter typically produces a more representative subsample but is much more expensive and labor-intensive.
Of particular concern are methods that require small masses, such as the 1 g typically used for metals digestion. Increasing the initial mass to a minimum of 10 g at a <2-mm sample particle size improves reproducibility (Clausen et al. 2018a). There are generally only two options to reduce FE: increase the sample size to be analyzed or reduce the particle size. For typical soil and analyte concentrations of 1 ppm, to reduce FE to ≤15%, either the sample mass must be increased to 32 g (2-mm particle size), or the particle size must be reduced to less than 325 mesh (0.044 mm) for a 1-g sample.
An important element to consider when using a subsampling process is that the final subsample mass must be used completely in the analytical sample preparation step to maintain subsample representativeness. For this reason, the final target mass for each of the following approaches and the mass needed for analytical sample preparation must be considered when choosing the process.
5.2.6 TCLP
TCLP (USEPA Method 1311) is a method-defined parameter. Some ISM sample processing options have limited application to samples intended for TCLP analysis – for example, sample drying is not mentioned in Method 1311, so additional laboratory-based drying should not be applied to samples intended for TCLP since it might affect analyte stability or leachability. In addition, limited particle size reduction might be needed to meet the <9.5-mm particle size criterion. Two-dimensional slabcake subsampling on the sample can be used to produce 5- and 100-g subsamples for the pretest and leaching steps.
Sometimes, optimal processing options differ for different analytes. In these instances, there are three options: (1) collect separate ISM field samples for each option, (2) split the field sample with different portions going to the different processing options, or (3) collect analytical subsamples at different points in the sequence of processing options.
Collecting separate field samples is the statistically preferred option but also the most expensive. Splitting the field sample prior to processing is inexpensive but can introduce significant reproducibility and uncertainty issues because the splitting process occurs before any ISM processing.
The more cost-effective and reasonably reliable option is to collect analytical subsamples at appropriate points in the processing sequence. The most common intermediate subsampling points and the reasons for choosing them are listed in Table 5-2.
Table 5-2. Subsampling points in the ISM processing sequence.
Source: ITRC ISM Update Team, 2020.
| Subsampling Point | Reason to Choose | Limitation |
| As-received sample when initially spread out for 2D slabcake prior to air-drying | Low boiling point, weakly sorbed analytes might be lost during air-drying | Small-scale heterogeneity is likely to be high |
| After disaggregation | No particles are to be excluded from the subsamples | Small-scale heterogeneity is likely to be moderate |
| After sieving | Need to avoid milling because of potential analyte loss | Small-scale heterogeneity is likely to be small to moderate |
Often times, the collection of an ISM sample yields a kilogram or more of sample material, but splitting the bulk sample in the field or laboratory is generally not recommended to reduce the sample mass because the entire sample should be processed. The uncertainty introduced by splitting prior to the completion of sample processing can be large when the COPC nugget effect is large, such as in highly heterogeneous samples. Note that bulk sample splitting (or subsampling) without particle size reduction merely increases FE.
Paired ISM sample collection is generally recommended over bulk ISM sample splitting when different sample processing treatments will be needed. Paired ISM samples allow the application of such treatments without the uncertainty introduced by bulk splitting.
5.3 ISM Sample Preparation and Analysis
Sample preparation options include moisture management, disaggregation, sieving, milling and subsampling. Analysis includes sample digestion or extraction and instrumental analysis methods suitable for the matrix and COPCs.
5.3.1 Moisture management
To facilitate air-drying, place the soil sample on a tray made of or lined with a material compatible with the COPC (see air-drying tower in Figure 5-5). The selection of the tray or liner material should ensure that the analytes of interest are neither lost nor gained from the sample to the tray or liner by sorption or reaction, and potential interferences are also avoided. Aluminum trays and liners should be avoided if aluminum is a COPC or if it may interfere or interact with an analyte of interest (such as chromium or elemental mercury). Plastic trays and liners should be avoided if phthalates and plastic components are COPCs, and a paper liner should be avoided if organic carbon or organics that can sorb to paper (such as petroleum) are COPCs. Spread the sample evenly in the drying tray, and if needed, use 2D slabcake subsampling to collect a subsample for moisture determination of the original sample. Place the sample in a ventilated area such as a hood or oven with sufficient airflow to carry away evaporated moisture. Drying time varies from a few hours to several days depending on moisture content, soil characteristics, airflow, and temperature. Intermittent (for example, daily) turning of the soil may be necessary to facilitate air-drying in an acceptable time frame. The soil should be dry enough to allow the agglomerates to be crushed producing a flowable matrix; moisture content below 5 to 10% is usually acceptable. Wet clay samples should be crushed with a pestle partway through the drying process to avoid the formation of large bricks that are difficult to handle with subsequent processing. Drying to a constant weight is not necessary; the sample only needs to be dry enough to facilitate proper mechanical function of subsequent processing equipment. The ventilated air-drying area uses a large amount of laboratory space during the drying step, so the use of racks to hold the drying trays can facilitate an efficient use of space. Air-drying towers with filters on the front and fans on the back have been effective at consistently and quickly air-drying samples. Drying via ovens at elevated temperatures or high-speed forced air is generally not recommended.

Source: Mark Bruce, Eurofins, 2019. Used with permission.
Disaggregation. To disaggregate, take the dry sample and gently rub it into a sieve to promote break-up of the soil agglomerates. A variety of sieve sizes can be used depending on the project DQOs, but a #10 sieve (2 mm) is the most common size. Alternatively, the soil can be disaggregated using a bladed coffee-type grinder or blender, just keep the time as short as possible to minimize wear on the blade, contamination of the sample with the blade materials, and any sample temperature elevation. A mortar and pestle can also be used to gently break up the soil agglomerates, though there is a greater risk of causing particle size reduction of the hard particles than with softer disaggregation techniques such as pestle/sieve and blender. Disaggregation is generally sufficient when SVOC COPCs are the primary concern and subsample sizes are 10 g or larger. Disaggregation and sieving are also commonly used prior to complete particle size reduction using the milling techniques listed in Section 5.3.3.
Other disaggregation tools include spinning blade coffee mills and food processors. A rotary hammer disaggregator breaks up soil clods by hitting them with a cylindrical rod while the soil falls through the path of the hammers (Figure 5-6).
5.3.2 Sieving

Source: Mark Bruce, Eurofins, 2019. Used with permission.
Particle size selection can occur at several different points in the ISM process. However, it generally follows air-drying and disaggregation with the 2-mm (#10) sieve being the most common (Figure 5-7).
Samples with little vegetation and composed mostly of sand and silt (which naturally have a very low moisture content) and air-dried soils can be sieved (typically using a #10 sieve, <2-mm particle size) in the field to remove pebbles and vegetative debris. Prior to air-drying or sieving or both, the field-moist sample weight should be recorded if specified in the SAP. The <2-mm particles are generally considered soil, while larger particles are considered gravel, rocks, or other materials (sticks and roots). Additionally, field sieving is an option that allows the user to calculate the mass of a bulk ISM sample needed to meet DQO requirements based on soil particle size. Unless field subsampling will be performed, the entire sieved ISM sample fraction should be submitted to the laboratory for appropriate processing and subsampling.

Source: Mark Bruce, Eurofins, 2019. Used with permission.
5.3.3 Milling
Extended high-speed milling can elevate sample temperature due to friction, so COPC thermal stability and volatility should be considered when choosing equipment and a milling scheme. USEPA SW-846 Method 8330B for nitrocellulose-based propellant residues specifies a 2-min (or longer) cool-down period between five 60-sec grinding intervals with a puck mill to maintain acceptable temperatures and minimize the loss of volatile energetic COPCs. The hard nature of explosive and metal particulates necessitates a 1-min milling interval.
When milling has been selected as part of the ISM DQOs, the entire conditioned ISM sample is milled. Splitting an un-milled ISM sample with high heterogeneity due to the nugget effect can lead to non-representative subsampling (Clausen et al. 2018a). If the milling equipment is not large enough to process the entire sample, mill smaller portions of the sample and then re-combine and mix them after the milling step. The milling equipment listed below is not an exclusive list of equipment capable of meeting ISM DQOs, but it is an example of equipment that has been used successfully in the past.
Mortar and pestle grinding can be accomplished with either manual or automated systems, with large automated systems recommended because of their increased capacity, better reproducibility, and reduced likelihood of repetitive-stress injuries. The sample contact materials can be steel, ceramic, or others depending on the COPCs. The sample is loaded into a heavy walled bowl, then crushed between the bowl wall and the pestle by manually pushing the pestle or spinning the bowl with a fixed pestle in an automated system.
Rotary pulverizers can reduce particle size from approximately 6 mm to <100 μm, the distance between the grinding plates determines the final particle size. The dry sample is fed into the chute, and the ground sample is collected from a hopper beneath the grinding plates. Note that adequate cleaning of rotary pulverizers between samples to remove any potential cross-contaminants is difficult.
Ball mills consist of both high- and low-speed systems. Typically, the sample is placed in a container along with a grinding medium and shaken rapidly or tumbled slowly. The milling medium (typically steel or agate balls, or ceramic cylinders) crushes the sample particles. High-speed systems consist of high-strength containers and high-speed shakers, which means they can provide more reproducible reduction to <100-μm particle sizes. Typical milling time for high-speed systems is a few minutes.
Low-speed systems typically consist of single-use cans, a milling media, and a low-speed tumbler or roller. Roller mills or paint can shakers are common examples. Typical milling times are several hours, but excessive milling should be avoided due to possible analyte loss. A study of anthropogenic metals in particulate form demonstrated a minimum milling interval of 18 hrs (Clausen et al. 2012).
Dish and puck (shatter box) milling are described in USEPA SW-846 Method 8330B (USEPA 2006c). The sample is loaded into the dish with the puck inserted. If the dish is not large enough to process the entire sample at once, mill smaller portions of the sample and then re-combine and mix after the milling step. The milling cycle time and cooling period (if necessary) depend on the analytes of interest. An example cycle consists of 1 min of milling and at least 2 min without milling to allow the dish and sample to cool. This process may be repeated two to four more times, depending on the materials to be milled. The cooling part of the cycle reduces internal temperatures and hence thermal degradation of the analytes. USEPA SW-846 Method 8330B (energetics) recommends a final particle size of <75 μm, but the optimal milling conditions and final particle size for other COPCs might be different from that guideline. Performance for other COPCs should be demonstrated with reference materials or other known samples.
Sieving can also be used to determine whether the milling step is complete. Particles below the DQO-specified size are removed from the milling process, and those above the predetermined cutoff size are returned to the milling equipment for additional particle size reduction. Common maximum particle size cutoffs are 250 μm (#60), 150 μm (#100), and 75 μm (#200). Alternatively, final particle size suitability can be estimated by touch or visual inspection when less accuracy is acceptable.
5.3.4 Sample mixing
Dry mixing can reduce heterogeneity and facilitate representative subsampling if the sample consists of particles of similar size and density. However, mixing samples with large differences in particle size or density can increase stratification and hinder representative subsampling. Unless, particle size reduction is performed, dry mixing is generally not recommended.
5.3.5 Subsampling (2D slabcake on dry or wet samples, rotary sectorial splitter)
The preferred subsampling methodology is the 2D Japanese slabcake or incremental sampling approach that emulates the field incremental subsampling process in a controlled laboratory setting. The entire sample is spread evenly onto a 2D surface at a depth easily penetrated by a square scoop. A scoop is then taken by removing an increment that equally represents the entire vertical column of the slabcake, and the material is placed in a receiving container. This process is repeated at least 30 times at systematic random locations around the entire sample
The laboratory default should be to use at least 30 increments to build the analytical subsample. If project-specific planning has determined that other increment numbers are needed to meet the DQOs, then use them, but replicate subsamples are recommended to determine whether the subsampling meets the DQOs.
A process should be established to document that the increments are collected from random or systematic random locations over the entire exposed surface to ensure adequate representation of the sample. To do so, increments for replicate samples should be collected from independent locations, or alternatively, the entire sample may be stirred, re-spread, and replicate increments collected in the same manner as the primary sample, with the process repeating for as many replicate samples as applicable.
A good example setup is a 20- × 30-in aluminum baker’s tray lined appropriately. The tray can easily take a 2-kg sample spread across it at a depth of no more than 1 to 2.5 cm. A scoopula pushes the sample around and spreads it to an even depth, ideally, as thinly as practical. As the sample is spread, the fine particles tend to migrate downward toward the tray while the larger, less-dense ones rest on top. A scoop is then used to minimize the discrimination of taking more of the large particles on the top. A square-walled, blunt-end scoop with a minimum 16-mm width tends to perform the best because it facilitates equal collection from both the top and bottom of the slab (Figure 5-9); the sides reduce the tendency of particles to fall off the scoop during increment collection. Before taking increments, the target mass should be considered because each scoop (increment) will ideally represent <1/30th of the desired target mass – for example, for a 30-g subsample, each increment should weigh about 1 g. Before starting the scooping process, a few trial scoops should be taken and weighed to calibrate the amount needed for each scoop. This technique works best when used after disaggregation or milling in conjunction with particle size selection via sieving to reduce the range of particle sizes (Figure 5-8).

Source: Mark Bruce, Eurofins, 2019. Used with permission.
The 2D slabcake subsampling process may be applied to moist “sticky” samples as well. The best results are achieved with sieved soils, but this process can also be applied to as-received samples. Spread the moist soil into an even-depth slabcake as described above, then use a square-walled, blunt-end scoop with a minimum 16-mm width (Figure 5-9) to collect 30 or more increments of 2-mm particle size for the final analytical subsample. Coring tools may also be used for subsampling if a moist sample is sufficiently cohesive. See the tool width discussion in Section 4.4.
A 1D slabcake is produced by pouring the sample into a line and using at least 20 passes back and forth to distribute the sample particles over that line. A square scoop is cut across the line to remove a subsample aliquot, and the sampler can combine as many of these aliquots as needed to produce the target subsample size (Gerlach and Nocerino 2003).
Sectorial sample splitting is the statistically preferred process because it results in the least sample heterogeneity of the methods discussed, but it does require investment in a rotating sample splitter and dust-abatement measures. The device consists of a rotating head with several chutes sitting on top of a motor. The chutes are spaced equally apart from each other and are of the same dimensions. A hopper is mounted above the rotating head with a vibrating tray that delivers the soil sample to the splitter at a variable rate, depending on the intensity of the vibrations. The rotation speed should be adjustable. The sample falls from the hopper into the chutes as they spin, and collection devices such as sample bottles are mounted on the bottom of each chute to receive equal portions of sample material. In general, slower feed rates from the hopper and faster rotational speed make for better subsamples.
The entire sample must be poured into the hopper initially, with the resulting subsamples equal in mass to the initial sample mass divided by the number of subsamples. If the desired target mass is not achieved on the first split, re-combinations of individual splits may be required to achieve a larger final target mass or re-splits of one of the previous spilt samples (serial splitting) if a smaller mass is needed. Small amounts of fine particles may adhere to the device and should be pushed through by tapping or by a small burst of compressed air. Limitations to this technique include equipment cost and availability, trained staff availability for correct operation, equipment cleaning issues, and equipment maintenance.
Riffle splitting generally divides the sample into two equal portions by directing the sample portions into opposite pans with alternating chutes. It can be used sequentially to further subdivide a sample into smaller aliquots (Gerlach and Nocerino 2003). Riffle splitting has been demonstrated to introduce a high degree of error in samples, however, and is not recommended for the collection of subsamples for analytical testing (Pitard 2019).

Source: Mark Bruce, Eurofins, 2019. Used with permission.
If field subsamplingis to be performed, the entire ISM sample should be air-dried (only if necessary) and sieved to the predetermined particle size (typically using a #10 sieve, <2-mm particle size). The remainder of the processing steps are the same as those described in the 2D slabcake section earlier.
The mass of sample required for the analytical test or tests helps determine the mass of each of the 30 or more increments – for example, if a mass of 30 g is required for analytical extraction and analysis, 30 separate 1-g increments would be collected from systematic random locations. Depending on project DQOs, replicates of the field processed soil should be collected and submitted for analysis to evaluate the precision of the ISM field processing procedure. The entire submitted subsample mass must be prepared for analysis due to possible particle size discrimination during sample transit (such as fines settling to the bottom of the sample container). If the entire contents of the submitted container are not to be analyzed, the laboratory must use proper techniques to ensure a representative particle size subsample is used for analysis. Laboratory replicates should be analyzed to evaluate the precision of the laboratory’s subsampling procedure.
Simply dividing an ISM sample (sieved or not) into separate volumes and placing each volume into separate sample containers for analysis is not an acceptable method of mass reduction. Likewise, manually mixing samples to homogenize them in the field or laboratory may just serve to further segregate different particle sizes because particles may settle in layers by weight or size during mixing. The process of spreading the entire sample out in a thin layer and collecting many representative increments in a systematic random fashion with a tool that can scoop to the bottom of the sample is the best way to collect a representative subsample of all the different sizes and types of soil particles present.
Finally, if ISM sample processing and subsampling is performed in the field, it is recommended that at a minimum three replicate subsamples be collected and submitted to the laboratory for each different analysis. The subsampling (as described above) process should be repeated on oneISM sample to form replicates, and these replicate results can then be used to evaluate the precision of the field processing and subsampling. Note that, the subsampling replicates should be collected in addition toISM field replicates.
Limitations to the field processing of ISM samples include the following:
- It is not recommended for COPCs deposited as solid particulates (energetics, metals at firing ranges, and so on).
- It requires a controlled environment to air-dry, sieve, and subsample, if necessary, to minimize the potential loss or introduction of COPCs during processing.
- Additional subsampling replicates are needed to evaluate precision.
- More knowledgeable/trained field personnel required.
5.3.6 Sample processing of VOC ISM samples
ISM samples can also be collected for VOCs for contaminant analyses. ISM VOC sampling procedures should minimize soil disturbance and possible VOC loss due to volatilization by using methanol field preservation. Refer to Section 4.2.5 for additional details on the field collection of VOC samples and precautions (hazardous material handling and shipping) for methanol field preservation. Collection is based on the high-concentration method as described in Sections 2.2 and 8.2.2 of USEPA SW-846 Method 5035A (USEPA 2002f), with a minor modification to the sample container (bottle) to accommodate the increased number of increments (soil mass) and methanol volume per ISM sample. Typically, a coring device (Figure 5-10) and larger narrow-mouthed amber bottles (500 to 1,000 mL) with Teflon-lined caps (Figure 5-11) are required for ISM VOC sampling.
| ISM samples can be collected for VOC contaminant analyses, with ISM increments placed directly into the appropriate volume of methanol in the field. |
Typically, the bottle and solvent are prepared and pre-weighed at the laboratory prior to shipment to the field to allow for laboratory calculation of the final ISM soil mass. The volume of solvent should at least equal the mass of soil that will be introduced, and the headspace to preserved sample ratio (methanol + sample) should be less than or equal to that commonly achieved with discrete methanol VOC preserved samples (say, 32 mL headspace to 8 mL preserved sample). Appropriate surrogate compounds should be added to the sample bottle containing the methanol prior to sample collection, if appropriate to meet project-specific needs. Details should be specified in the SAP, and any alterations due to unforeseen field conditions should be recorded in field logs. When target analytes require immersion in a solvent, trip and field blanks (no sample added) should be included, depending on the DQOs. For example, when sampling for VOCs, if samples are immersed in methanol in the field, then trip blanks and field handling blanks – that is, bottles containing methanol – should travel to and from the field, and the field blank bottle(s) should be opened in the field under the same conditions and for the same amount of time as the sample bottles. Close coordination with the analytical laboratory regarding ISM VOC bottle/preservation requirements, sample kit preparation, sample receipt requirements, and so on is essential.
U.S. Department of Transportation regulations limit air shipment of containers with more than 30 mL of methanol (DOT 2019). As an alternative, a 60-mL bottle with 25 mL of methanol can be used to collect five 5-g increments, then the methanol extracted from the appropriate number of small bottles can be combined to represent the entire DU. A five-increment small bottle facilitates working with 30-, 50-, and 100-increment DUs. Alternate containers with flexible sides to minimize headspace during sampling are in development and can be considered when commercially available.

Source: Courtesy www.ennovativetech.com.

Source: ITRC ISM-1 Team, 2012.
A minimum of a 1:1 ratio of solvent volume to sample soil mass (that is, 1 mL of methanol to 1 g of soil) is recommended. This is a conservative recommendation, since a 5-g plug of soil typically has a volume of around 3 mL. Soil increments should remain completely submerged at all times, so additional solvent may be required to ensure that the sample mass meets this requirement (this requirement should also be discussed with the laboratory). The sample container should be selected based on the total mass of soil to be collected and the solvent required – for example, for 30 increments of 5 g each will require approximately 3 mL volume of solid material per increment, so a minimum of 150 mL solvent is recommended (Figure 5-11). The container should be large enough to accommodate additional solvent (if needed) and to prevent loss of solvent through splashing as soil increments are dropped into the container. The headspace to preserved sample ratio (methanol + sample) should be less than or equal to that commonly achieved with discrete methanol-preserved VOC samples. Potential headspace loss in ISM VOC samples is expected to be comparable to conventional discrete methanol-preserved VOC soil samples (see USEPA SW-846 Method 5035A) (USEPA 2002f).
Additionally, a separate unpreserved soil sample for percent moisture determination should be collected, if necessary, to report the ISM VOC results on a dry-weight basis. Typically, the unpreserved soil sample is collected in the same manner as the ISM VOC samples, meaning a second increment is collected at each ISM increment location, placed in an unpreserved wide-mouth container (4 oz or larger), and submitted to the laboratory.
The following equipment and information are necessary for laboratory processing and analysis of ISM VOC samples:
- The bottle tare weight (including sample label) and volume of methanol must be documented to back calculate the soil mass in the submitted ISM VOC sample. The density of methanol (0.7918 g/cm3) should be used for the calculation.
- The laboratory must have an analytical balance capable of weighing the ISM VOC sample as received.
- A separate unpreserved soil sample, collected in the same manner as the preserved ISM VOC sample, should be submitted for percent moisture determination.
- If required, total volume and moisture correction should be performed by the laboratory for final contaminant concentration reporting, per Section 11.10.5 of USEPA SW-846 Method 8000C.
Typically, a 24-hour period is sufficient to extract VOCs from most soils. Tight clays are an exception and may take several days (Fehsenfeld et al. 1992). Therefore, caution should be taken if the plugs of soil do not readily disperse when submersed in methanol. Soils should be completely disaggregated or dispersed in the solvent to ensure efficient extraction.
A potential drawback of ISM for VOCs is that the methanol preservation (high-concentration method) approach results in lower sensitivity. Specifically, the methanol dilution step causes elevated analytical detection limits, method detection limits (MDLs), reporting limits (RLs), practical quantitation limits, and so on compared to the direct soil purge-and-trap, low-concentration method’s techniques. Analytical detection limits could be elevated above relevant screening levels for certain targeted contaminants. If the analytical detection limits (or other issues) present difficulties in using ISM for VOCs, this issue should be discussed with the laboratory and the overseeing regulatory agency prior to sample collection. If the projected analytical detection limits are too high to be of use or some other issue restrains the use of these methods at a specific site, alternative approaches may need to be used. Options may include alternate analytical methods/techniques (such as SIM) to achieve lower detection limits or select discrete sampling via USEPA SW-846 Method 5035A (low-level VOC sampling).
5.3.7 Sample digestion or extraction
Analysis of energetics uses the solvent acetonitrile for extraction from the soil sample, with a small portion of the acetonitrile extract analyzed by chromatography, usually using SW-846 Method 8330 (USEPA 1994). Typically, a 10-g subsample built from 30 increments of the milled material is extracted with 20 mL of acetonitrile. Walsh and Lambert (Walsh and Lambert 2006) found acetonitrile extraction on a shaker table was equivalent to using acetonitrile in an ultrasonic bath. Other organic analysis subsamples are usually in the 10- to 30-g range.
Metals analysis with Method 3050B involves a standard digestion mass of 1 to 2 g using nitric acid to recover the environmentally available metals and hydrogen peroxide to remove organics. Improved precision is evident with larger digestion aliquots (Clausen et al. 2018c). Consequently, for metals analysis, it is recommended that 10 g of material be obtained during subsampling and subsequently digested if the sample has not been milled.
Subsamples smaller than 10 g tend to have more variability but still might meet project objectives. Collect an initial subsample of 10 to 30 g and split the subsample into the target subsample mass (<10 g) using miniature versions of 1D or 2D slabcake.
5.3.8 Analysis
Method 8330 specifies using High Performance Liquid Chromatography with an ultraviolet detector, which has been the most widely used analytical approach for detecting energetic compounds in soil samples from military sites. Another method (USEPA 1999b) employs the same sample processing steps as Method 8330 but uses a gas chromatography with an electron capture detector for determination. There is no reason that this method of determination could not be used with the sample processing steps specified in Method 8330B.
5.4 Quality Assurance/Quality Control
The overall quality assurance program provides the structure and contains the specific quality control elements used to meet project DQOs.
5.4.1 Introduction
Regardless of the COPCs, it is imperative that sample processing and analysis include appropriate QA and QC measures. QA/QC requires careful upfront planning during DQO development (see Section 3.1) and is dependent upon the intended use of the data. In addition, practitioners must also consider any corrective actions and/or decision rules that will be followed in the event that any QA/QC milestones are not met. Most of the QA/QC criteria applied to laboratory ISM procedures evaluate representativeness of the processed laboratory subsample, analyte loss, or cross-contamination during processing. Therefore, it is emphasized that failure to adhere to processing procedures of samples will affect the ability to meet project quality goals. Users should first have a firm understanding of the differences between QA and QC.
5.4.2 DQOs and laboratory coordination
As outlined in USEPA DQO guidance (USEPA 2006b), the DQO process is used to establish the performance and acceptance criteria that serve as the basis for designing a plan for collecting data of sufficient quality and quantity to support the goals of the study. The project delivery team must decide during the initial project planning phase which of the sample processing and analytical options currently available and applicable to ISM are most appropriate to achieve the ISM project DQOs. This includes sample conditioning steps, such as drying; disaggregation; particle size reduction (if warranted) via milling, crushing, or other means of particle size selection using sieving to focus on a particle size fraction of interest; and finally what analytical subsampling techniques and/or determinative analytical methods will be performed. QA/QC can also be affected if the processing is not in a controlled environment – for example, if any sample processing such as sieving, drying, or subsampling occur in the field.
The project delivery team will determine the project needs for sampling and sample processing as documented in the QAPPs. Before award, the laboratory must review the QAPPs (including all QA/QC criteria) and appropriately determine if they can meet project needs, so close interaction of the team or the project chemist with the laboratory is critical here. Specifically, although the processing, preparatory, analytical methods, and procedures needed to meet the DQOs are predetermined and documented in the QAPPs, the laboratory will compare in-house capabilities to project QA/QC and determine if they can meet those project needs. It is also critical that the QAPP document any corrective actions that will be necessary in the event that any DQOs are not met – for example, will sample reanalysis be required if any surrogate or grinding blanks fail performance criteria?
The following sections discuss various options regarding sample processing and analysis for ISM. It is imperative that close communication and coordination with the analytical laboratory take place from the initial project planning phase and DQO formulation through ISM sample collection and subsequent sample processing and analysis to ensure that defined data of known quality and usability are obtained for the project. The DQOs, including QA/QC criteria, are always planned for and documented in the project QAPPs.
5.4.3 Quality assurance
Choosing the specific laboratory processes to handle and analyze incremental samples is influenced by the specific COPCs, the nature of the release, and the objectives of sampling event. For example, COPC volatility and thermal stability affect the options for sample conditioning, while exposure scenarios and the objectives for risk assessment may influence choices related to particle size reduction. In addition, sample drying and particle size reduction can create a negative bias due to analyte loss. Biologically degradable analytes with very high boiling points may remain stable when the sample is air-dried, but some of the processing procedures may need to be modified or avoided entirely if a COPC is sufficiently volatile and/or biodegradable. Negative bias created by target analyte loss may also significantly impact attainment of project DQOs, and as these considerations apply to certain volatile metal species, they require careful control to maintain acceptable milling temperature. Negative bias due to analyte loss can affect MDLs and precision as well, so in all cases, the laboratory selected to perform the sample processing and analysis should have empirical data showing that each ISM standard operating procedure (SOP) has been appropriately validated (including the potential affects from all sample processing steps) for each analyte being evaluated and, where possible, in representative matrices. A project chemist should compare laboratory SOPs to the project’s QA/QC requirements as documented in the QAPPs.
5.4.4 Quality Control
Monitoring for bias due contamination or analyte loss is accomplished with blanks and the use of known concentration spikes or samples. Precision is monitored with replicates.
5.4.4.1 Monitoring cleanliness and carryover
ISM sample processing can result in a positive bias due to sample cross-contamination of equipment – for example, milling equipment can contribute metal concentrations to the sample through the milling, crushing, or pulverizing apparatus. Common metals include chromium, cobalt, iron, manganese, nickel, and tungsten, with metallic composition analysis and guidance usually available from the manufacturer (such as avoid high chrome steel when low ppm concentrations of chromium are of interest). Other malleable metals, such as lead or copper, may smear in milling machinery. If a significant amount of larger particle size, malleable metals are expected in ISM samples, additional sieving and fractional analysis should be considered, or alternative sample preparation techniques may need to be investigated.
With each of these subsampling techniques, consideration should be paid to the potential for contamination. Decontamination processes must be developed and checked using a matrix such as blank Ottawa sand at an established frequency between samples. The composition of the subsampling equipment should also be considered as a potential contamination source. Plastic parts containing phthalates, for example, should be avoided if SVOC phthalates are COPCs.
Sample preparation blank. Sometimes referred to as a method blank, a sample preparation blank is a clean sample matrix that, when available, is used to establish whether equipment used to process samples (dry, pulverize, split, mix, and so on) has been adequately cleaned between field samples. The sample processing blank is used to assess whether there is carryover or cross-contamination during the processing steps. The blank should be evaluated by milling and analyzing a clean matrix immediately after preparing a sample of known or suspected high malleable metal content, but note that clean soil matrices are more likely to be available for organic analytes. For metallic analytes, no known soil-like matrices are available that will produce ultra-trace (non-detect results) at environmental levels of concern for all likely metals. Again, the project team should work directly with the laboratory to first determine underlying project needs, then establish the tolerable levels of potential carryover/cleanliness. Method blanks should go through all the phases of sample preparation, subsampling, extraction, and analysis that are experienced by field samples.
5.4.4.2 Monitoring bias
Laboratory control sample (LCS). The LCS is a known matrix spiked with compound(s) representative of all target analytes. It represents a “best case” control of overall method performance as it is performed on a clean matrix spiked with the COPCs. It is also used to document possible analyte loss and/or overall laboratory method performance. LCS control limits must be established by the laboratory for each ISM procedure and analysis performed, and then provided in the final laboratory report. Usually, there is one LCS sample processed and analyzed per batch. As the LCS is intended to evaluate overall method performance, it should experience the same processing steps as that of field samples.
However, many laboratories have introduced the LCS after ISM sample processing because it is costly to add all target analyte spikes of sufficient concentration to a 1-kg or greater ISM sample. This might be acceptable on a project-specific basis, but the potential bias caused by sample preparation procedures prior to LCS processing must be evaluated by other means (such as surrogates). LCS acceptance criteria are typically generated by each laboratory using appropriate statistical evaluation of multiple LCS samples, but monitoring the air-drying and sieving steps may be problematic for many organic compounds. The deposition of the spiking solution onto the LCS might not result in a sample with spiking compounds bound in the same manner as the sample contaminants themselves. Moreover, the association between low boiling point SVOCs and the clean soil or sand matrix might be significantly weaker than in weathered field samples. Thus, potential losses from an air-dried laboratory prepared control sample can be significantly higher than from a field sample.
Theoretically, it is also possible that COPCs are bound significantly to the matrix and will not be dissociated completely during the extraction/digestion step, but these same compounds will be easily extracted/digested in the LCS. However, if, for example, air-drying, sieving, and subsampling are the only ISM sample processing steps being performed, a “standard” (that is, approximately 30-g) clean matrix spiked LCS carried through all these steps would present the potential “worst case” analyte loss to be evaluated for some analytes and the “best case” analyte recovery to be evaluated for other analytes. The best case/worst case scenario for the LCS exists for discrete samples, too. The typical sample size of an ISM sample is 1 kg, but an LCS may not need to be that same size – it may only require the same preparation and analysis process.
Synthetically fortified soils may not produce the same strength of interactions between the COPCs and the soil particles. In particular, QA/QC materials spiked at the laboratory or other commercial providers may overestimate contaminant losses during ISM sample processing steps. Reference materials from weathered “native” contaminated soils are more likely to match the loss rates for field samples; standard reference materials (SRMs) are currently available for explosives (USEPA 2006c), as is a 500-g solid commercially available QA/QC standard for energetics. Such material is often analyzed as an LCS on a per batch basis, but other analyte groups might become available over time.
Project-specific DQOs should be assessed during systematic planning to determine the appropriate analysis frequency. Nitrobenzene, 2-nitrotoluene, 3-nitrotoluene, and 4-nitrotoluene have low recoveries when the QA/QC standard is air-dried at room temperatures, so the DQO process needs to address whether the QA/QC standard will be air-dried or only milled. There can be significant costs associated with a commercial QA/QC standard, but a separate QA/QC standard is available for tetryl (an energetics constituent) and should be considered if it is a target analyte. The frequency at which a QA/QC standard needs to be processed and analyzed should be defined during the SPP, depending on project-specific DQOs. With respect to energetics, additional guidance for laboratory QA/QC can be found as part of USEPA SW-846 (USEPA 1999b) and “DOD Quality Systems Manual for Environmental Laboratories (QSM)” (DOD/DOE 2018).
ISM samples collected for non-volatile metals may also include drying, sieving, and milling preparation steps. It is assumed that this process does not cause the loss of metal analytes, so it may not be necessary to require a large-scale LCS throughout the entire process. The necessity for a metals LCS (large-scale or otherwise) should be defined during the SPP, depending on project-specific DQOs.
Monitoring the effectiveness of the milling step for metals, explosives, or other particulate-based analytes is best demonstrated by adding these analytes in solid particulate form (such as metal salts), rather than the traditional liquid spike solutions used by laboratories. Demonstrating the ability to produce representative subsamples from heterogeneous samples would require the original QA/QC sample to be intentionally heterogeneous and not the highly homogenized reference materials commonly available from providers.
Much of the focus of this QA/QC section is targeted on the milling portion of the ISM process, largely due to the paradigm shift from conventional sample preparation and analyses. Simply put, particle size reduction is an invasive sample handling technique and therefore requires an additional level of QC. Note that for some organic analytical methods using ISM, particle size reduction may not be necessary. The primary purpose for milling is reduction of FE by reducing the particle size and eliminating larger nuggets that can be the cause for extreme in-sample heterogeneity.
Matrix spikes. MS are intended to evaluate any potential intrinsic matrix properties of the field samples that might affect sample extraction and analysis. An MS is an aliquot of an actual field sample spiked with a known concentration of target analytes, and as the intended objective of the MS is to evaluate matrix issues rather than ISM processing, the spiking occurs just prior to extraction and analysis. Due to the bulk mass spiking limitation, modifications may be necessary for MS analysis. For sites with a large degree of heterogeneity, it may be necessary to collect a duplicate ISM sample to use with this type of MS approach so as not to remove a portion of the primary ISM sample because it could ultimately bias the representativeness of the analyzed sample result to that of the original sampled area. MS/MS duplicates (MSD) might not be needed for precision assessment if subsample replicates are used.
Surrogates. A surrogate is used in the preparation and analysis of samples for organic analytes in order to evaluate the potential bias introduced during sample processing. In this case, surrogates are added to the entire sample as received from the field and go through all the same sample preparation and analysis steps. The surrogates used should be similar to the target analytes and are typically isotopically-labeled analogs of the target compounds of concern. Unlike LCS and MS, which are performed once per batch and in a clean matrix, surrogates are added to each sample, blank, and LCS prior to any processing steps, and the resulting recoveries allow the user to evaluate any potential bias experienced by that specific sample. As previously discussed, an ISM field sample received for analysis must be processed as received in its entirety. Because ISM samples are typically 1 to 2 kg in mass, there are logistical and financial challenges that come with using surrogates with each ISM sample:
- Laboratory costs significantly increase as additional waste is generated. In addition, the concentration of the surrogate fortification stock must be appropriate in order to achieve a final concentration within the linear range of the calibration after sample processing, subsampling, extraction, and analysis.
- Depending on the corrective actions chosen in the event that QC recoveries fail performance criteria, multiple field replicates may need to be collected if the corrective actions require samples to be reprocessed. In addition, multi-method surrogate additions to the sample field replicate may not be feasible as surrogates from one method may interfere with the analysis of another method – for example, surrogates used for Method 8270 interfere with some PCB, organochlorine pesticide, and petroleum hydrocarbon analyses. This may also add to the costs incurred for the collection, transport, and storage of the additional samples.
- Standard recommended surrogate recoveries are provided in each respective organic method in USEPA SW-846, but these method-specific criteria do not include ISM-specific sample preparation procedures (such as drying and milling). Therefore, method-specific surrogate recoveries are not recommended, and each laboratory should define their own performance criteria based on empirical data during method validation.
- As previously mentioned for LCS fortification, because of the difference in the strength of interactions between weathered soil particles and contaminants, surrogates spiked at the laboratory or other commercial providers may overestimate contaminant losses during ISM sample processing steps. Therefore, stable surrogates pre-fortified onto solid spike materials are recommended, rather than simply spiking the surrogate as a solution onto the field sample.
5.4.4.3 Monitoring precision
Laboratory replicates. Laboratory replicates (also known as subsampling replicates) are recommended to assess the precision of ISM subsampling processes and ensure that the subsample selected for analysis is representative of the entire field sample collected for processing. Three subsample replicates are recommended on samples selected by the project team with a targeted RSD as determined during the project-specific SPP. Generally, replicate subsamples of 30 to 50 increments each should be collected after all ISM processing is complete. These replicates should then be carried through the rest of the analytical process. A 15 to 30% RSD for laboratory replicate precision is generally considered confirmation that the processed sample is sufficiently homogenized. The frequency of these laboratory replicates can vary from one replicate set per batch to one set per project, depending on the project DQOs. Therefore, including appropriate subsampling procedures and monitoring subsampling replicate precision is especially critical where sample drying and particle size reduction have not been performed. Historically, analytical processing precision has been evaluated via MS and MSD recoveries, but evaluation of this precision through the evaluation of laboratory subsampling replicates is a superior metric when the analytes are present at sufficient concentrations.
5.4.4.4 Other QC considerations
It is often desirable to perform sample splitting (obtaining identical splits of a field sample post sample collection) in order to send the splits to separate laboratories. Paired ISM sample collection (or field replicates) is generally recommended over bulk ISM sample splitting when different sample processing treatments will be needed. Paired ISM samples allow separate sample processing procedures to be conducted without the uncertainty introduced through bulk splitting. Moreover, the error introduced by splitting prior to the completion of sample processing can be large when the COPC nugget effect is large, such as in highly heterogeneous samples. Note that bulk sample splitting (or subsampling) without particle size reduction merely increases FE.
To help ensure data quality, all field sampling, field processing, and laboratory sample processing activities should be supervised by personnel trained in ISM. Samples should be shipped to an accredited laboratory following recommended protocols for the class of target analytes, and laboratory SOPs for ISM sample processing and analysis should be requested and reviewed by the project chemist. Chain-of-custody, field notes, and data validation reports should also be retained and utilized in the final data usability assessment (DUA).
QC measures should be implemented both in the field and laboratory. When sample processing and subsampling are initiated in the field to reduce the amount of sample shipped off site, replicate samples of the processed soil should be taken to establish the uncertainty introduced by this step (see Section 2.6.2.1). It should be noted that reducing the mass of the sample shipped to the laboratory will tend only to increase FE. Depending on the contaminant, field blanks and/or equipment blanks also may be required. Field blanks are often necessary for VOCs and some SVOCs, particularly when a solvent is involved.
Laboratory accreditation/certification. Project teams must be aware of the accreditation requirements that apply to their projects because such requirements may vary based on the program and state under which the sampling is being performed. They may also vary based on whether the procedure follows a formal published method, is based on a formal published method, or is an internal laboratory procedure. In most systems, accreditation is given at the fields of testing (FOT) level, where each combination of matrix (such as non-potable water, drinking water, and solid and chemical materials), method/technology, and analyte is considered an FOT.
Three primary types of accreditation requirements exist:
- The National Environmental Laboratory Accreditation Program (NELAP) is a national program implemented by member states, with state governmental agencies usually serving as accreditation bodies for state-selected programs and FOTs. A NELAP accreditation body will accept by recognition the accreditation status of a laboratory issued by another NELAP accreditation body (called secondary accreditation). For more information, see www.nelac-institute.org.
- Each state has its own procedures to address accreditation of method modification and internal laboratory procedure. Some states have elected not to participate in NELAP, and some have retained separate accreditation structures for certain programs. Each of these states has its own procedures to address accreditation of method modification and internal laboratory procedure.
- Some federal agencies have their own accreditation programs. The U.S. Departments of Defense and Energy (DOD and DOE) have centralized a Environmental Laboratory Accreditation Program (ELAP). The DOD ELAP program has specific ISM quality requirements listed in the “DOD Quality Systems Manual” (DOD/DOE 2018) that cover laboratory QC requirements for samples collected via incremental sampling. In addition, the DOD QSM provides ISM-specific quality requirements for explosives by 8330B (QSM table B-3); ISM samples being analyzed for parameters other than explosives should utilize QSM table B-23.
Click Here to download the entire document.